The immune system constitutes a complex network of biological processes that serves to safeguard organisms from various diseases. An important role of the immune system is its ability to distinguish between normal cells in the body and foreign cells such as germs and cancer cells. This discrimination empowers the immune system to target foreign agents while preserving normal cellular integrity. If the immune system malfunctions, it mistakenly attacks healthy cells, tissues, and organs which is called autoimmune disease. These attacks can affect any part of the body, weakening bodily function and even turning life-threatening. Therefore, it is very critical to maintain a balance between combating tumor cells and avoiding autoimmune disease in the body. The immune system maintains such balance by using checkpoint proteins on immune cells. The checkpoints act like switches that need to be turned on (or off) to ensure an optimal immune response. However, cancer cells sometimes find ways to use these checkpoints to avoid being attacked by the immune system.
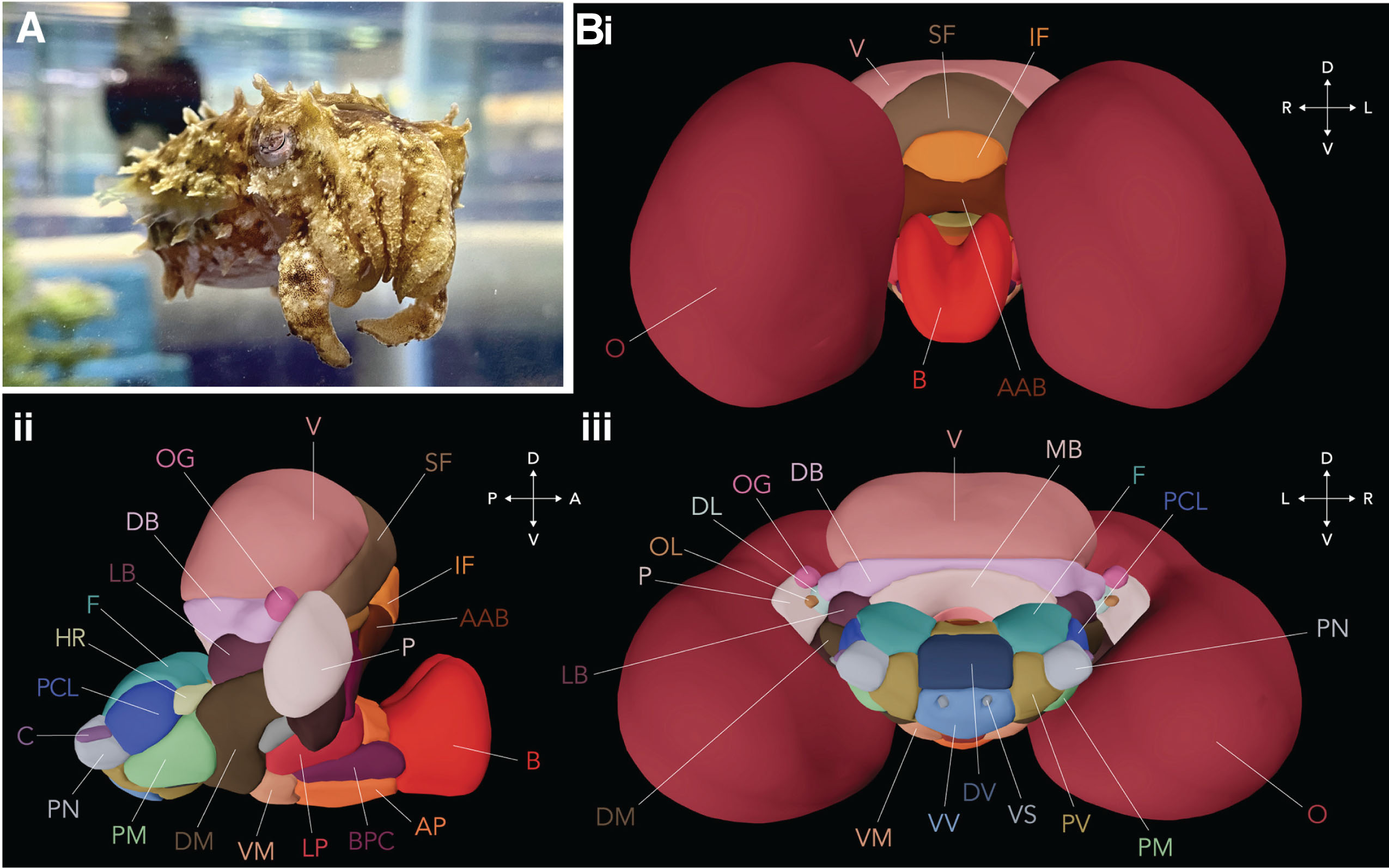
Medicines known as monoclonal antibodies can be designed to target these checkpoint proteins. These drugs are called immune checkpoint inhibitors. Checkpoint inhibitors don’t kill cancer cells directly, but they help the immune system to better find and attack the cancer cells in the body. Immune checkpoint inhibitors such as anti-PD-1 antibodies, are now used to treat some types of cancers such as metastatic melanoma or brain tumors due to their ability to boost the immune response against cancer cells. It normally acts as a type of “off switch” that helps keep the T cells from attacking other cells in the body. It does this when it attaches to PD-L1, a protein on some normal (and cancer) cells. When PD-1 binds to PD-L1, it basically tells the T cell to leave the other cell alone. Some cancer cells have large amounts of PD-L1, which helps them hide from an immune attack (Figure 1).
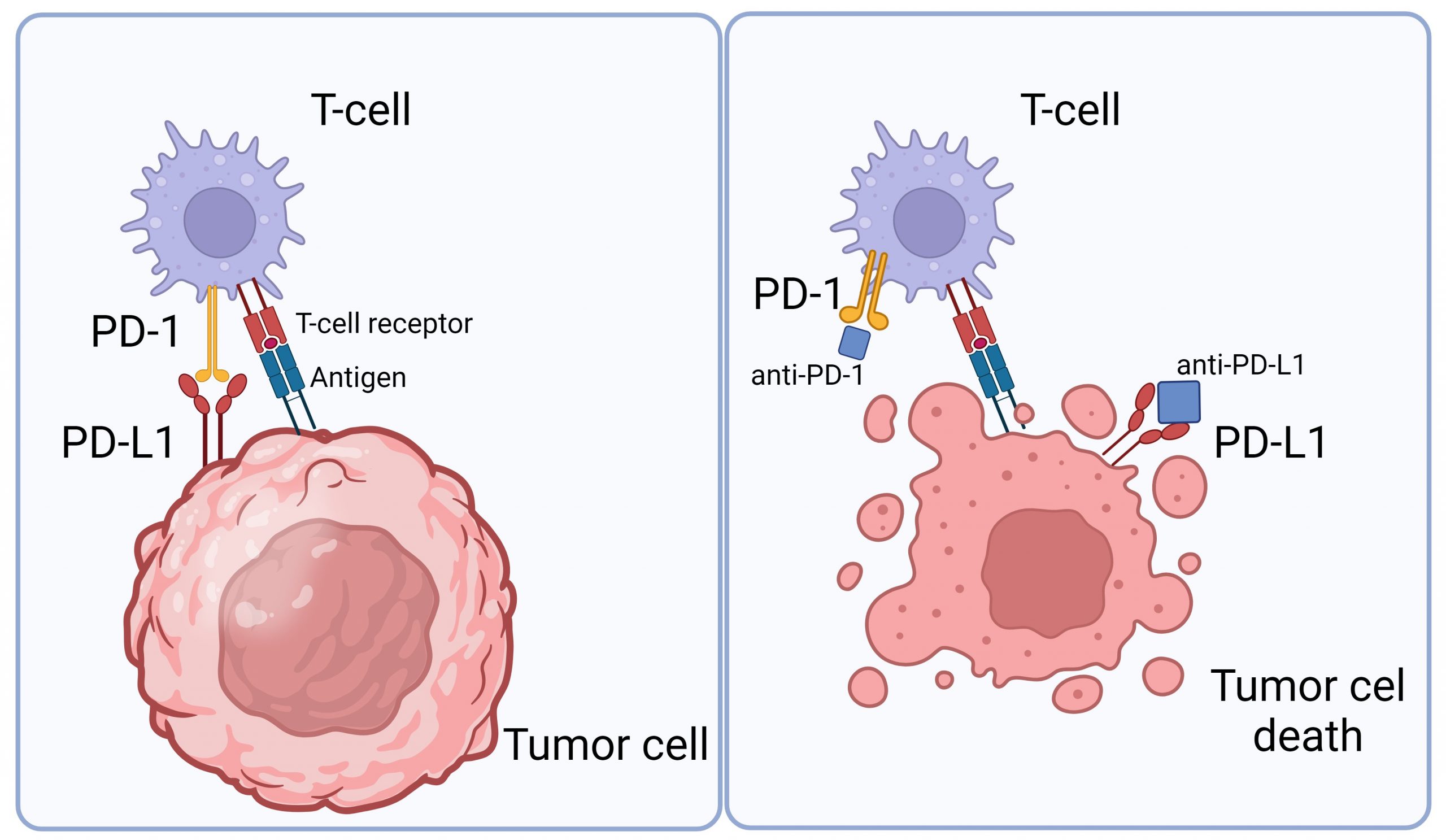
Figure 1: The interaction between tumor cells and T cells. Tumor cells contain PD-L1 receptors that bind to PD-1 in T cells to hide an immune attack from T cells (left panel). However, through the application of an immune checkpoint inhibitor (anti-PD-L1 or anti-PD-1), this binding is hindered, empowering T cells to eliminate tumor cells (right panel). Image is created via Biorender.
The Izar group at Columbia University Medical Center, led by Patricia Ho identified a mechanism by which CD58 loss or downregulation contributes to cancer immune evasion and immune checkpoint inhibitor (ICI) therapy. CD58 is known as an adhesion molecule facilitating the initial binding of effector T cells. By sequencing the whole genome of patient samples, specifically those who underwent anti-PD-1 antibody treatment and later developed drug resistance, they illuminated the role of CD58. Notably, CD58 facilitates early T-cell infiltration into tumors within a tumor differentiation. Moreover, the study shed light on CD58 loss potentially expediting tumor progression by upregulation of PD-L1, consequently intensifying resistance to ICI therapies.
To better understand the CD58/PD-L1 co-regulation, the research group performed a genome-wide knock out screen to identify the regulator of CD58. Among the top hits of cell population with low CD58 protein, authors identified CMTM6 as an important regulator for the T cell activation and antitumor responses. CMTM6 binds to CD58 and promotes its recycling from cell surface to endosomes, thus preventing its lysosomal degradation (Figure 2). Interestingly, CMTM6 is also known as an important regulator of PD-L1 maintenance. Thus, loss of CD58 results in increased PD-L1 levels by stabilizing CMTM6-PD-L1 interactions and therefore reducing lysosomal PD-L1 degradation (Figure 2).
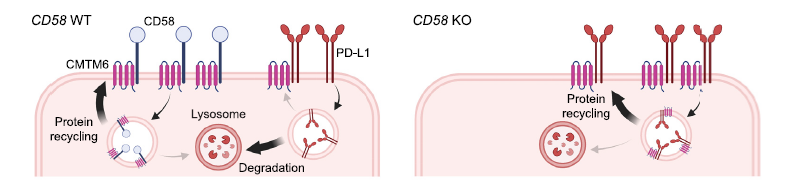
Figure 2: Both CD58 and PD-L1 require and directly bind to CMTM6. CD58 forms a direct binding with CMTM6, enhancing recycling and curtailing lysosomal degradation (left). CD58 knockout fosters PD-L1 up-regulation by interacting with CMTM6 (right). Figure is adopted from the original paper.
This work provides a molecular basis for clinically relevant roles of CD58 to facilitate initial infiltration into the tumor. Loss of CD58 confers resistance to immune checkpoint inhibitors which may provide important insights and inform rationale for the next generation of combinations of cancer immunotherapies.
Read more about this novel finding here: The CD58-CD2 axis is co-regulated with PD-L1 via CMTM6 and shapes anti-tumor immunity.
Reviewed by: Martina Proietti Onori and Carlos Diaz Salazar Albelda