For decades, scientists have known that the fast solar wind, a steady stream of charged particles from the Sun, comes from coronal holes, dark regions in the solar atmosphere where magnetic field lines open into space. But the origin of the slow solar wind, a more variable and less understood component of the Sun’s particle flow, has remained a puzzle. Now, a new study published in The Astrophysical Journal by Columbia postdoc Alexandros Koukras and his colleagues suggests that the boundaries of coronal holes may hold the key. Their findings not only provide the most detailed measurement to date of elemental abundances (how different chemical elements are distributed in the solar atmosphere) at these transition zones, but also offer an unexpected solution to the so-called missing open flux problem, where measurements of the Sun’s magnetic field in space don’t match what models predict at the solar surface.
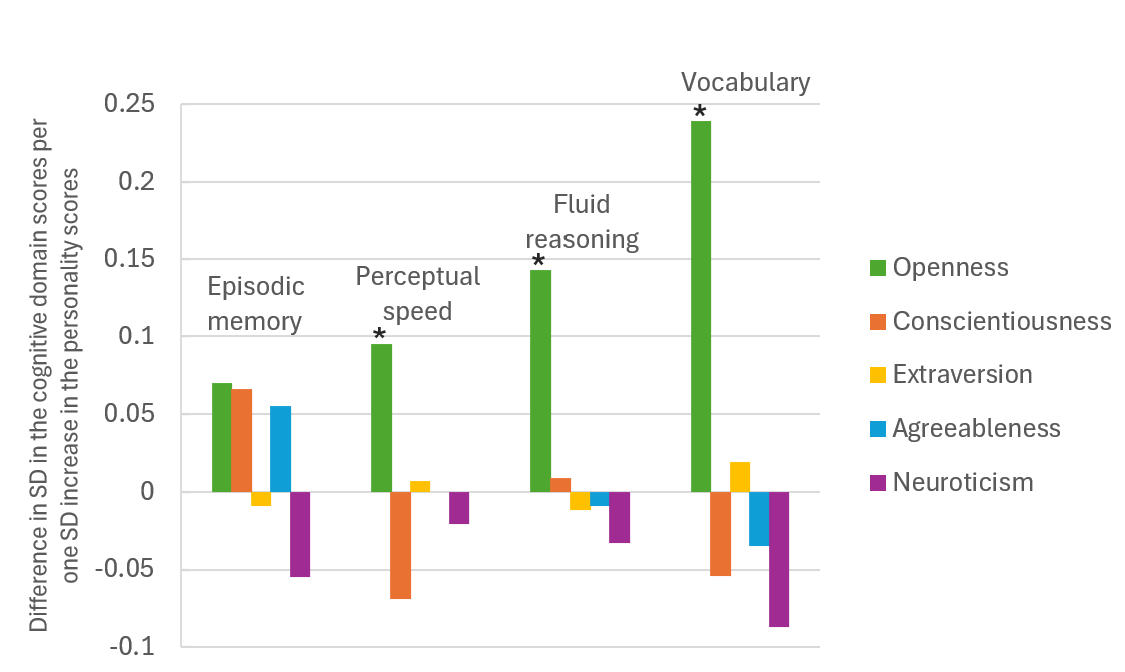
The Solar Wind’s Slower, Stranger Sibling. Unlike the fast solar wind, the slow wind appears to come from plasma that was once trapped in closed magnetic loops and then somehow escaped. The most likely escape route? A process called interchange reconnection, where open and closed magnetic field lines “swap” connections, freeing previously confined material. This process is thought to happen at coronal hole boundaries (CHBs), the turbulent edges where open-field coronal holes meet the magnetic loops of the quiet Sun. The “quiet Sun” refers to the vast, relatively stable regions of the solar surface that are neither active regions nor coronal holes. These areas are filled with dense magnetic loops and contribute significantly to the Sun’s background radiation and structure. But until now, it’s been difficult to detect direct evidence of this process in action. The study focuses on exactly this type of transition zone. As shown in Figure 1, a dark coronal hole sits at the center of the Sun’s disk, flanked by brighter quiet-Sun regions. The research team measured a clear gradient in elemental composition across the outlined boundary area: direct evidence of interchange reconnection in action. Though visually subtle, these edges are dynamically rich zones where magnetic field lines open, plasma escapes, and the slow solar wind may be born.

Figure 1. Extreme ultraviolet (EUV) image of the Sun captured by NASA’s Solar Dynamics Observatory on March 25, 2016. The dark area at the center is a coronal hole, a region where magnetic field lines open into space (lower brightness indicates lower density and temperature). The bright surroundings represent the quiet Sun, filled with closed magnetic loops. The blue box highlights the region analyzed in this study (located at the boundary between the coronal hole and the quiet Sun) where open and closed magnetic field lines interact through interchange reconnection. In this transition zone, researchers detected gradients in plasma composition, revealing magnetic reconnection in action. Adapted from the original paper.
Reading Magnetic Fields Through Chemistry. To investigate this, Koukras and his colleagues turned to a powerful diagnostic: the first ionization potential (FIP) bias. This is the ratio of an element’s abundance in the corona compared to the photosphere, and it’s known to change depending on whether plasma is confined or freely escaping. Using extreme ultraviolet (EUV) spectroscopy data from the Hinode spacecraft, the researchers measured how the FIP bias changes across a CHB. They applied two independent analysis methods: differential emission measure (DEM) modeling and linear combination ratios (LCR), to validate their results. What they found was striking: a clear, consistent gradient in the FIP bias extending outward from the coronal hole boundary, across a region roughly 30-60 megameters wide, about the size of a supergranule on the Sun. (A megameter is equal to one million meters.)
A Hidden Source of Solar Wind and Open Magnetic Flux. These gradients aren’t just chemical quirks. They trace ongoing magnetic reconnection. As field lines shift from closed to open, they carry enriched plasma into space, fueling the slow solar wind. But the study also revealed something even more surprising. By calculating the amount of magnetic field “leaking” through these boundary regions, the researchers estimate that 37–71% more open magnetic flux may originate from CHBs than previously accounted for. This is a big deal, because models of the Sun’s open magnetic field consistently underestimate what spacecraft measure in space. The discrepancy is known as the open flux problem, and this new study suggests that we’ve simply been overlooking an important source.
Small Loops, Big Impact. Another interesting twist? The reconnection at these boundaries seems to involve smaller magnetic loops than those typically assumed in models. While previous theories focused on massive 200 megameter loops, this study shows that reconnection can happen with loops as small as 30-60 megameters. This has implications for how we model solar dynamics, especially during active periods in the solar cycle. This research offers one of the clearest observational insights into interchange reconnection at the solar surface. It bridges local plasma behavior with global heliophysical questions, such as how the solar wind is generated and how magnetic energy is transported throughout the space surrounding the Sun. Future missions like Parker Solar Probe and Solar Orbiter may help confirm these findings with in-situ data. But already, this work points to a new understanding of how the quiet edges of the Sun contribute to its most dynamic outputs. For more detail, check out the original publication.
Reviewed by: Maithê R. M. de Barros