The population is aging. Over the past two decades, life expectancy in the United States has increased by more than five years to approximately 80 years in 2020 and is projected to further increase to over 85 years in 2060. The progressive rise in life expectancy leads to a growing share of older people in society, which increases the demand for health care services including home health care (HHC). HHC is defined by medical services that are provided in a patient’s home, usually by skilled nurses. An important aspect of HHC is the identification of patients who are at high risk for emergency care. A substantial fraction of HHC patients has to visit the emergency room or is admitted to the hospital during the HHC period. Strikingly, up to 30% of these events could be avoided by accurate risk prediction and preventative care. For example, patients identified as high-risk can be monitored more closely and treated with additional medications to prevent adverse health outcomes.
The research of Columbia postdoc Jiyoun Song is aimed at improving risk prediction in the setting of HHC. In recent work published in the Journal of Advanced Nursing, Dr. Song and colleagues performed cluster analysis, an unsupervised machine learning method to aggregate available patient data into groups. Such a clustering method is useful in identifying patterns of risk factors that interact with each other, rather than examining individual risk factors. The analysis was performed on structured data from electronic health records, that include patient characteristics such as socio-demographics and health conditions, as well as unstructured data from clinical notes written by nurses. The approach of Dr. Song and colleagues to use data derived from these clinical notes is quite innovative. They extracted risk factors from these notes through the artificial intelligence (AI)-based tool of natural language processing. Natural language processing can be used to extract meaning from text written by a human, which forms the foundation of AI chatbot systems such as the recently developed ChatGTP. The use of clinical notes as an additional source of data significantly improves risk prediction in the HHC setting, as previously shown by Dr. Song.
Through AI-assisted cluster analysis of both the structured and unstructured data, three clusters of risk factors were identified with distinct characteristics: (1) impaired physical comfort with pain, (2) high comorbidity burden (i.e., a person is suffering from more than one physical disease or condition at the same time), and (3) impaired cognitive/psychological and skin integrity. Classifying patients in these three categories could help tailor individualized preventive strategies. For example, a pain management strategy may work best for patients in cluster 1, while patients in cluster 3 may benefit mostly from psychological counseling and wound management. The study also found that these clusters are associated with the frequency and timing of emergency room visits. Patients that fall into cluster 3 have the highest need for emergency care, with 15.7% of patients being hospitalized or visiting the emergency department within the first 60 days of HHC (see Figure below).
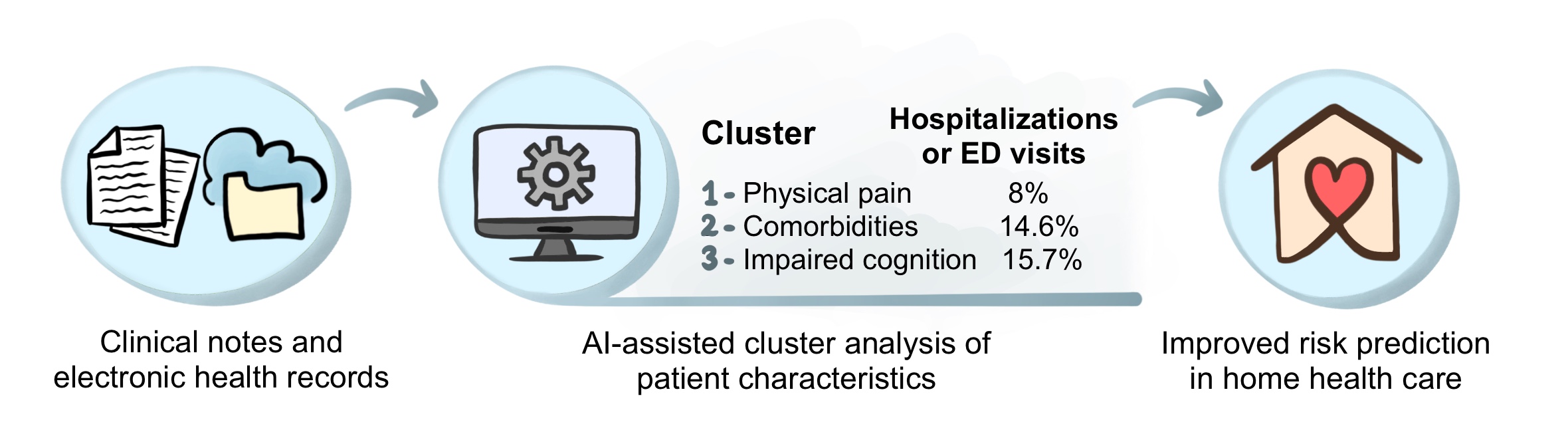
AI-assisted cluster analysis of both structured data (i.e., electronic health records) and unstructured data (i.e., clinical notes) identified three clusters of risk factors with distinct characteristics. The risk of hospitalizations or emergency department (ED) visits is different for each cluster. Such a cluster-based analysis is useful for identifying high-risk patients in home health care and implementing preventative strategies. © 2022, Maaike Schilperoort
The results from this study suggest that implementation of cluster-based risk prediction models into early warning systems could reduce the likelihood of HHC patients being admitted to the hospital. This illustrates the potential of AI-based methods for clinical risk prediction. The use of AI is getting increasingly popular and is now also implemented in various other aspects of healthcare, such as in-hospital decision making, predicting treatment benefit, and personalizing medicine. To what extent will AI be incorporated in our healthcare system? Only time will tell.
Reviewed by: Jiyoun Song, Pei-Yin Shih, Trang Nguyen, and Sam Rossano