Imagine It’s a Saturday morning and you decide to drive upstate for a nice hike. While on the trail, you have a too-close encounter with a too-curious bear, leaving you panting, perplexed. This acute stress response (the famous fight-or-flight response) is extremely useful for us to better react to danger. The main hormones released during a stress response, glucocorticoids (GC), help prepare our body for the adverse situation we may encounter, giving us a better chance of survival.
Chronic stress it’s not so beneficial for us. In fact, it has profound detrimental effects on our bodies, and especially in our brain. There is increasing evidence that it can make our mitochondria (the powerhouse of the cell) function poorly, which can lead to damage in our neurons. Another way chronic stress can damage the brain is by inducing accumulation of the protein Tau, which can lead to neurodegenerative diseases such as Alzheimer’s disease. Tau itself poses an interesting biological conundrum: While it is normally found in the neuron’s microtubules (the cell’s scaffolding) in a healthy setting, it can start to form clumps (i.e, oligomerize) when a neuron becomes stressed or damaged, interfering with the neuron’s basic functions such as synaptic transmission and protein transport.
Scientists are still unsure how exactly GC cause Tau to oligomerize. Furthermore, while the impact of GC in mitochondria’s fitness is pretty well studied, the mechanisms driving such dysfunction, and its downstream consequences, are still poorly understood. Overall, to the scientific community, the molecular mechanisms linking GC exposure, mitochondrial dysfunction, and Tau pathology, are still unclear.
In a recent study published in Brain, Columbia postdocs Dr. Fang Du and Dr. Qing Yu and colleagues investigated the causal relationship between these three important components of brain health, and found that GC directly precipitate mitochondrial dysfunction and drive Tau oligomerization. In a series of elegant experiments in both mouse models (in vivo) and neurons grown in petri dishes ( in vitro),experiments, the group led by Dr. Clarissa Waites demonstrated that found the mechanism by which GC exposure drives mitochondrial dysfunction and Tau oligomerization, and even discovered a potential treatment.
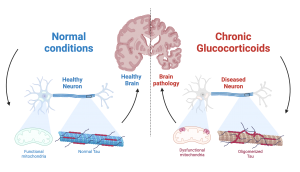
First, Dr. Waites’ group established the role of chronic GC exposure in mitochondrial fitness and pathogenic Tau accumulation in vivo. Using the widely used synthetic glucocorticoid dexamethasone (DEX), they demonstrated that GC induce mitochondrial dysfunction (increased permeability and lower respiratory capacity) and oligomeric Tau accumulation. Next, Drs. Du and Yu very cleverly employed in vitro experiments to demonstrate that DEX triggered mitochondrial dysfunction by increasing mitochondrial permeability: their research showed that GC exposure stimulates the opening of the mitochondrial permeability transition pore, a big, non-discriminant, pathological hole in the mitochondrial membrane that when open, greatly compromises its function. Most importantly, they found that GC triggers opening of the pore by activating its key component, the protein cyclophilin D. This key finding led Dr. Waites’ group to block cyclophilin D activation, either genetically (i.e. changing the DNA of the protein) or using drugs, with great success: cells where cyclophilin D activation was prevented were remarkably resilient to GC stress, and showed much lower levels of mitochondrial dysfunction and pathological Tau accumulation.
Although this was an extremely exciting result by itself, it posed a problem moving forward: neither genetical nor pharmacological inhibition of cyclophilin D are currently viable therapeutic approaches, due to their poor bio-availability or invasive methods required to deliver the treatments. So what other approach could they use to also prevent mitochondrial pore opening, while still being clinically tractable? After some digging, Drs. Du and Yu found apo-cyanin, an orally-available inhibitor of mitochondrial respiration that could tackle GC-driven brain pathology while not directly targeting cyclophilin D. In line with their predictions, this drug was able to restore mitochondrial function and prevent Tau pathology in DEX-treated cells. Most importantly, they were also able to restore neuronal health, neuronal connectivity, and prevent anxious and depressive behavior associated with GC treatment.
All of these experiments clearly demonstrated that GC-exposure triggers mitochondrial dysfunction and Tau accumulation, which could in turn promote the development of brain pathologies. How could they demonstrate, however, that targeting mitochondrial permeability is a feasible approach to treat common brain pathologies such as Alzheimer’s disease? To tackle this question, Dr Waites’ group made use of an ingenious technique, where they replaced all mitochondria in a test cell with those coming from an Alzheimer’s disease patient. Excitingly, in this model, treatment with mito-apocynin was sufficient to partially reverse mitochondrial dysfunction and Tau oligomerization found in the cells containing mitochondria from Alzheimer’s patients.
Dr Waites’ group story is a hallmark out-of-the-box thinking, and clinical relevance. While their research clearly highlights mitochondrial damage induced by GC and cyclophilin D activation as a key trigger of Tau accumulation, the implications of this finding could be much broader: How generalizable is this mechanism to other forms of brain disease such as ischemia, inflammation, or aging? As a matter of fact, cyclophilin D levels are elevated in Alzheimer’s patients, and its inhibition is protective in mouse models of sclerosis, Parkinson’s, and Alzheimer’s disease.
While this story provided incredible insight into the role of mitochondria in controlling brain pathology, many other questions remain unanswered. For example, What is the exact mechanism by which mitochondrial dysfunction promotes pathogenic Tau accumulation? It is highly likely that these two events are intertwined, and that oligomeric Tau can also drive mitochondrial dysfunction.
Despite the still-unknowns, it is clear that after this publication from Dr Waite’s lab, we should re-contextualize our middle-school textbooks: The mitochondria is the powerhouse of the cell, and during stress, the gate-keeper of our brain’s health.
Reviewed by: Trang Nguyen, Erin Cullen