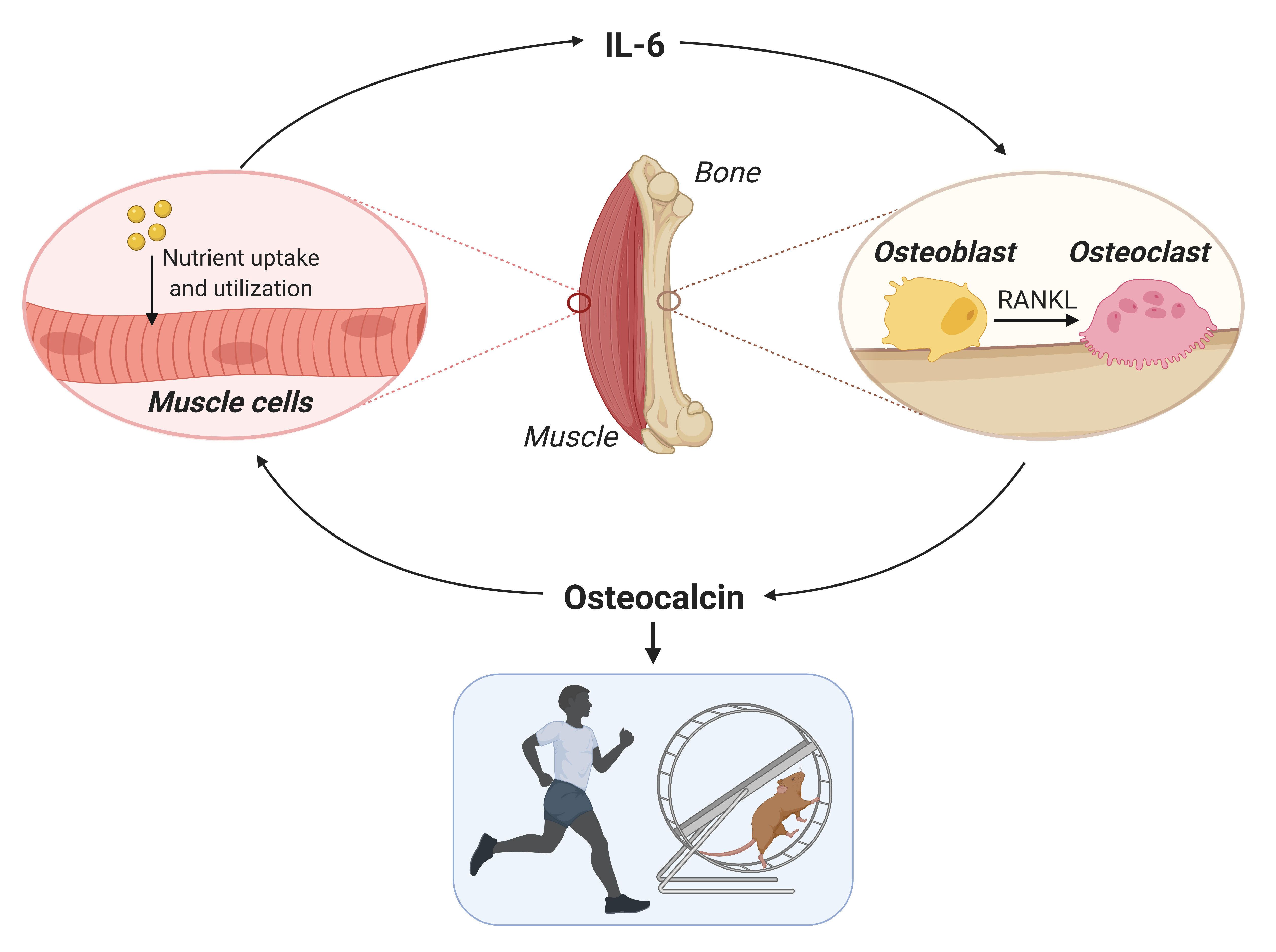
During your childhood, your parents might have added a sweet flavor to the bitter medicines that you did not want to take. Do you wonder why you were getting a bitter taste anyway? There is a scientific explanation.
Attraction to sweet compounds and the aversion to bitters are innate behaviors triggered by the mammalian taste system. Despite their apparent simplicity, the neuronal mechanisms that trigger these behaviors are highly complex. Alterations in the sense of taste are quite common in adults. The most common taste dysfunctions are loss (ageusia) or reduced (hypogeusia) sense of taste. Interestingly, ageusia is one of the most frequent symptoms reported after infection with COVID-19. Response to bitter and sweet taste starts when chemicals in the food activate specialized cells called taste receptor cells on the tongue and palate. These cells make contact with matching ganglion neurons, which form a bridge from the periphery to the brain. In the brain, bitter and sweet signals are represented by spatially distinct populations of neurons in the taste cortex that receive these signals through the brainstem. Scientists are investigating the brain regions and mechanisms that regulate this circuit and how bitter and sweet responses intermingle.
In a recent study in Cell, Dr. Hao Jin and colleagues uncover the regulatory mechanisms of neuronal responses to sweet and bitter taste in mice and how this modulation is important when sweet and bitter responses are combined. Aversion to bitter taste is well-recognized to be an innate behavioral response important to detect and prevent ingestion of harmful chemicals. So, how does the behavioral rejection of a bitter taste prevail even when combined with a sweet taste? To address this question, the authors firstly aimed to identify the neural population in the brainstem responsive to bitter and sweet taste. Dr. Hao Jin and colleagues tested the evoked response to artificial sweetener and bitter substances in subsets of neurons in the brainstem using fiber photometry, a prominent in vivo imaging technique that quantifies the neuronal activity of a region or a population of brain cells in awake animals. They found that a specific population of neurons (b-neurons for simplification) were specifically active in response to bitter tastes, while the activity of a distinct neuronal population (s-neurons) was enhanced solely after sweet stimuli. A series of experiments were then performed to functionally validate these neurons in the brainstem as a passage of response to bitter and sweet tastes. To start, it was observed that chemical ablation of b- or s-neurons leads to a decreased avoidance of bitter solutions and a loss of attraction to sweet stimuli, respectively. Additionally, the authors asked whether selective activation of b- and s-neurons in the brainstem was sufficient to evoke a taste response even without a taste stimulus. Using optogenetics, a technique that allows to artificially increase or decrease neuronal activity through light, Dr. Hao Jin and colleagues observed that activation of b-neurons in mice decreased licking to bitter substances while activation of s-neurons increased licking to sweet stimuli.
In addition, the authors asked why and how responses overlap and the reason for the bitter to overcome sweet stimuli. They observed that sweet taste responses from s-neurons were largely suppressed when a bitter stimulus is presented together with a sweet flavor. This suppression of sweet response was found to be directly executed by the taste cortex. Interestingly, at the same time, the activity of b-neurons is enhanced also by the cortex but via the central amygdala (Figure 1). As a result, despite the efforts of your parents to turn those bitter medicines yummy, a team of brainstem and central amygdala neurons are raising a red flag about the potential toxicity of the food you ingest (even if this is not the case), increasing the response to bitter taste and suppressing the response to sweet taste.
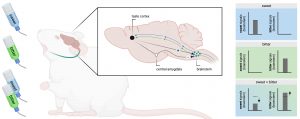
Figure 1. Schematic representation of the neuronal circuits involved in the response to bitter or/and sweet taste and how there are modulated. Adapted from Jin et al., and created with Biorender.com.
Dr. Hao Jin is a postdoctoral fellow at Dr. Charles Zuker ‘s lab in the Zuckerman Mind Brain Behavior Institute at Columbia University.