Remembering where we left our keys, the distance between the platform and the train, or where the TV remote control was placed last night, are examples of daily events that require spatial memory, i.e., the ability to recall spatial locations. This ability is made possible by neurons called place cells, which are mainly located within a brain region known as the hippocampus. While we navigate spaces, place cells encode information from environmental cues, creating a sense of space. Place cells were discovered by Dr. John O’Keefe, who, together with Drs. May-Britt Moser and Edvard I. Moser, was awarded with the Nobel Prize in Medicine and Physiology in 2014. The so-called ‘inner GPS’ of the brain is shared across species and has been identified in mammals including rodents, primates, and bats, which are particularly known for their spatial abilities.
In a recent article in Science, Columbia postdoc Dr. Payne and colleagues investigated whether this navigation system also exists in non-mammals with pronounced spatial memory skills. The authors chose to study tufted titmice, birds that accumulate seeds in multiple locations to prevent a shortage in case of decreased food supply. In the lab, the authors recorded hippocampal activity of tufted titmice while they were navigating in an arena for sunflower fragments. They identified neuronal activity similar to rodent place cells. Also, they observed a mammalian-like distribution of those cells along the hippocampus. These observations suggest that the mechanisms of hippocampal coding in birds exhibit similarities to those identified in mammals.
The authors compared results from the titmouse with the activity and hippocampal organization of zebra finch, a non-food-caching bird. They found that the subregion of the hippocampus with the highest density of place cells was larger and more populated in tufted titmice than in zebra finches. These findings suggest that place cells are more spatially informative and stable in tufted titmice compared to the place cells in zebra finches. Such differences in spatial coding between bird species might be due to differences in demands of food caching.
In this work, Dr. Payne and colleagues identified for the first time neuronal activity in a non-mammalian hippocampus similar to mammalian-like place cells. Furthermore, they reveal evidence that spatial encoding may differ between species mainly due to its functional demand. Despite a divergent evolution from millions of years, mammals and non-mammals possess a similar version of their GPS cells.
Dr. Hannah Payne is a postdoctoral fellow atDr. Dmitriy Aronov‘s lab in the Zuckerman Mind Brain Behavior Institute at Columbia University.
Acute lymphoblastic leukemia (ALL) is an aggressive form of cancer arising from malignant transformation of immature cells that were otherwise fated to become white blood cells, or lymphocytes. ALL occasionally affects adults but is more commonly a pediatric cancer: children under the age of 5 have the highest risk of being affected. Upon diagnosis, it is possible to treat ALL with an aggressive regimen of multiple chemotherapy drugs that is successful for over 80% of patients. Unfortunately, when tumors reappear after initial treatment, in relapse, the cases become extremely difficult to treat. Additionally, the cellular landscape of ALL in relapse has a high degree of genetic heterogeneity and variability between different patients. Oncologists and cancer biologists suspect it is this genetic complexity that makes ALL in relapse especially hard to fight in the clinic.
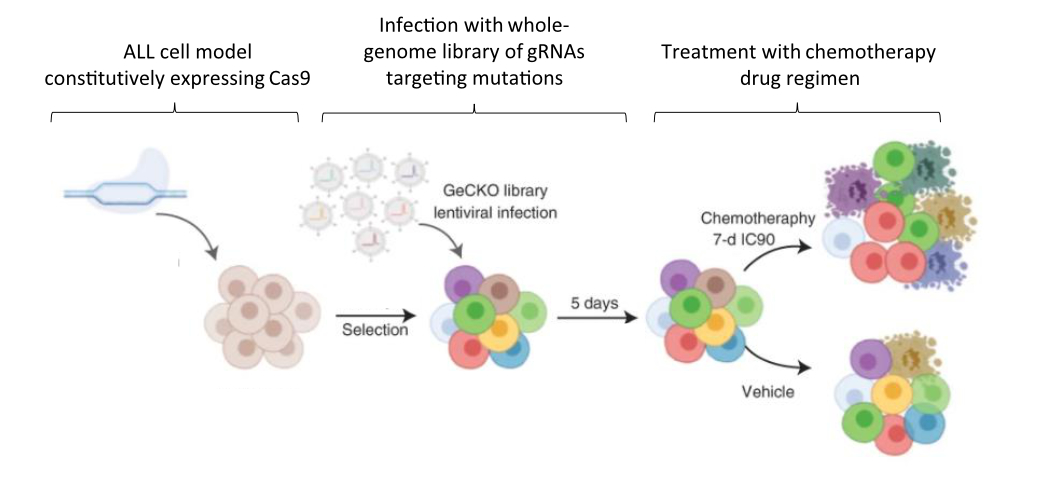
In a studypublished in Nature Cancer last year, Dr. Jessie Brown and colleagues set out to improve outcomes for patients with ALL by clarifying how mutational complexity in relapsed tumors interacts with chemotherapy drugs to resist treatment. To this end, the team first employed extensive genetic sequencing on ALL samples collected upon diagnosis, remission, and relapse in order to identify the mutational landscape that distinguishes relapse tumors from the others. Samples were collected from 175 patients in total – 149 from pediatric cases and 26 from adults. Next, in order to functionally characterize the mutations they identified, the authors used a genome-wide genetic screening strategy to identify drug–gene interactions and determine why the relapse-specific mutational landscape is less responsive to chemotherapy. The screen was carried out in a representative model ALL cell line using CRISPR, a genetic editing tool used to specifically activate or inhibit the expression of single genes.
Fig 1. Schematic of experimental design for CRISPR-based screen in ALL model cell line. A library of targeting molecules called guide RNAs (gRNAs) was used to activate or inhibit genes identified to have mutations associated with ALL in relapse. Figure adapted from Oshima, Zhao, Durán, Brown, et al. 2020.
Between these two approaches, the team succeeded in characterizing relapse-specific mutations that arise during the administration of chemotherapy itself, a process known as clonal evolution. Additionally, the number of mutations they identified increased with patient age at diagnosis, a finding that allowed the researchers to establish that the most recent commonality between the mutant cells present at diagnosis versus later at relapse often develops early, years before the leukemia is officially diagnosed. Importantly, this finding is consistent with the hypothesized fetal origin of many pediatric ALLs, which postulates that chromosomal abnormalities leading to cancer are already present at birth. It is also consistent with the higher rate of relapse previously observed in adult patients.
When the mutations uniquely acquired during relapse were further investigated using CRISPR screening in an ALL model cell line, a strong positive selection was revealed for those that conferred chemotherapy resistance. By using CRISPR to manipulate the expression of genes affected by each mutation of interest and assessing how the ALL model cells fared in each experiment, the researchers were able to analyze the relationships between the effect of the mutation and application of each drug. Of the drugs investigated, functional overlaps in the cellular mechanisms mediating the activity of each were identified between several groups of them. The significance of this finding is two-fold. First, it helps researchers and medical providers understand why the presently used multi-drug regimen might be effective for ALL in the first place. Second, it suggests that other drugs acting via similar mechanisms of action could be effective treatments in the future. Moving forward, ALL in relapse might be treated not just with combinatorial chemotherapy, but with specific combinations, doses, and schedules of drugs that meet the personalized genetic vulnerabilities of specific ALL cases.
One inhibitor tested in the study’s cell-based CRISPR screen, an inhibitor called ABT-199, also known as Venetoclax, is already being tested for inclusion as a new therapeutic. If approved, it could become part of the arsenal of drugs used to compose personalized chemotherapeutic cocktails for patients with ALL in relapse. According to co-first author Dr. Brown, “it is currently in Phase I/II clinical trials for relapsed ALL and other malignancies and we hope that this work and our follow-up studies can further underscore the mechanisms of action of this inhibitor in combination with commonly used chemotherapies.”
Altogether, this study identified a number of mutations that make relapsed ALL distinct from ALL before treatment with chemotherapy and functionally characterized the interactions of these mutations with multiple chemotherapy drugs. While ALL is not a common cancer – it accounts for less than 0.05% of all incidences of cancer in the United States – those affected must be treated with aggressive chemotherapy that can negatively affect the lives and health of patients in many ways. Because of this, understanding how to better target ALL at diagnosis and treatment-resistant ALL in relapse is a high priority for researchers. The findings of this work help to identify targets for reversing chemotherapy resistance and improving treatment outcomes for pediatric and adult patients alike.
Dr. Jessie Brown is a postdoctoral research fellow in the Ferrando Lab at Columbia University Irving Medical Center studying therapeutic resistance in relapsed acute lymphoblastic leukemia.
In empirical sciences, theories can be sorted into three categories with ascending gain of knowledge: empirical, semi-empirical and ab initio. Their difference can best be explained by an example: In astronomy the movement of all planets was known since ancient times. By pure observation astronomers could predict where in the sky a certain planet would be at a given time. They had the knowledge of how they moved but actually no clue why they did so. Their knowledge was purely empirical, meaning purely based on observation. Kepler became the first to develop a model by postulating that the sun is the center of the planetary system and the planet’s movement is controlled by her. Since Kepler could not explain why the planets would be moved by the sun, he had to introduce free parameters which he varied until the prediction of the model matched the observations. This is a so-called semi-empirical model. But it was not until Newton who with his theory of gravity could predict the planets movements without any free parameters or assumptions but purely by an ab initio, latin for “from the beginning”, theory based on fundamental principles of nature, namely gravity. As scientists are quite curious creatures, they always want to know not only how things work but also why they work this way. Therefore, developing ab initio theories is the holy grail in every discipline.
Luckily, in quantum mechanics the process of finding ab initio theories has been strongly formalized. This means that if we want to know the property of a system, for example its velocity, we just have to kindly ask the system for it. This is done by applying a tool, the so-called operator, belonging to the property of interest on the function describing the system’s current state. The result of this operation is the property we are interested in. Think of a pot of water. We want to know its temperature? We use a tool to measure temperature, a thermometer. We want to know its weight? We use the tool to measure its weight, a scale. An operator is a mathematical tool which transforms mathematical functions and provides us with the functions property connected to the operator. Think of the integral sign which is an operator too. The integral is just the operator of the area under a function and the x-axis.
The problem is, how do we know the above mentioned function describing the system’s state? Fortunately, smart people developed a generic way to answer this problem too: We have to solve the so-called Schrödinger equation. Writing down this equation is comparably easy, we just need to know the potentials of all forces acting on the system and we can solve it. Well, if we can solve it. It can be shown that analytical solutions, that means solutions which can be expressed by a closed mathematical expression, only exist for very simple systems, if at all. For everything else numerical approaches have to be applied. While they still converge towards the exact solution, this takes a lot of computational time. The higher the complexity of the system, the more time it takes. So for complex systems even the exact numerical approach quickly becomes impractical to use. One way out of this misery is simplification. With clever assumptions about the system, based on its observation one can drastically reduce the complexity of the calculations. With this approach, we are able to, within reasonable time, find solutions for the problem which are not exact, but exact within a certain error range.
Another way to find a solution for these complex problems is getting help from one of nature’s most powerful and mysterious principles: chance. The problem of the numerical exact solving approach is that it has to walk through an immensely huge multidimensional space, spanned by the combinations of all possible interactions between all involved particles. Think billions of trillions times billions of trillions. By using a technique called Random Walking the time to explore this space can be significantly reduced. Again, let’s take an example: Imagine we want to know how many trees grow in a forest. The exact solution would be dividing the forest into a grid of e.g., 1 square foot, and counting how many trees are in each square. A random walk would start in the forest center. Then we randomly choose a direction and a distance to walk before counting the trees in the resulting square. If we repeat this just long enough we eventually will have visited every square, therefore knowing the exact number, meaning the random walk converges towards the exact result. By having many people starting together and doing individual random walks stopping when their results deviation is below a certain threshold a quite accurate approximation can be obtained in little time.
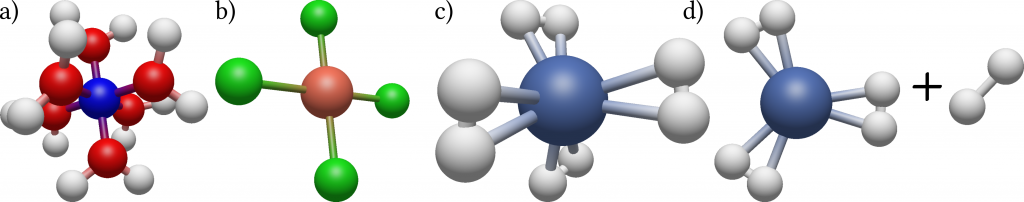
Columbia postdoc Benjamin Rudshteyn and his colleagues developed a very efficient algorithm based on this method specifically tailored for calculating molecules containing transition metals such as copper, niobium or gold. While being ubiquitous in biology and chemistry, and playing a central role in important fields such as the development of new drugs or high-temperature superconductors, these molecules are difficult to treat both experimentally and theoretically due to their complex electronic structures. They tested their method by calculating for a collection of 34 tetrahedral, square planar, and octahedral 3D metal-containing complexes the energy needed to dissociate a part of the molecule from the rest. For this, precise knowledge of the energy states of both the initial molecule and the products is needed. By comparing their results with precise experimental data and results of conventional theoretical methods they could show that their method results in at least two times increased accuracy as well as increased robustness, meaning little variation in the statistical uncertainty between the different complexes.
Figure 1: Illustration of the three types of geometry of the datasets molecules: Octahedral (a), square planar (b) and tetrahedral (c), with the transition metal being the central sphere. In (d) the dissociation of part of the molecule is shown.
While still requiring the computational power of modern supercomputers, their findings push the boundaries of the size of transition metal containing molecules for which reliable theoretical data can be produced. These results can then be used as an input to train methods using approximations to further reduce the computational time needed for the calculations.
Anyone who watched the movie Arrival would not miss the conspicuous resemblance of the alien Heptapods to some of our own earthly beings – the octopuses. While serving as inspiration for alien creatures in movies, as clairvoyants in soccer World Cup or as savages in classic science fiction and mythology, cephalopods like octopuses and squids have been dubbed as one of the most intelligent creatures on the planet. There is a good reason for why cephalopods, particularly octopuses, have developed such a reputation. Octopuses have a striking organization of brain structure, different from that of any other studied organism. They have the largest nervous system among animals lacking a backbone, comprising a total of nine “brains”. Out of these, one is a major donut-shaped brain that contains ~200 million neurons surrounding the octopus’s food pipe, which is strangely located in the head! This brain communicates behavioral intricacies to the eight so-called mini-brains located within the arms, each containing ~40 million neurons. The central brain is responsible for executing complex behaviors like tool use, ability to plan for the future, shape-shift, camouflage, recognize individuals and solve complex puzzles. While the last common ancestor between octopuses and humans was about 680 million years ago, a recent surprising discovery showed that they both evolved to use the same molecules during development. Scientists discovered that genes that produced the camera-like eye in humans are the same ones that gave rise to the camera-like eye in octopuses. What’s more, these cephalopods have evolved complex brains that show behavioral innovation on par with a small primate.
Comparison between the number of neurons present in the octopus and human brains. The octopus has one major brain and eight “mini-brains” while humans have neurons in the head and the spinal cord.
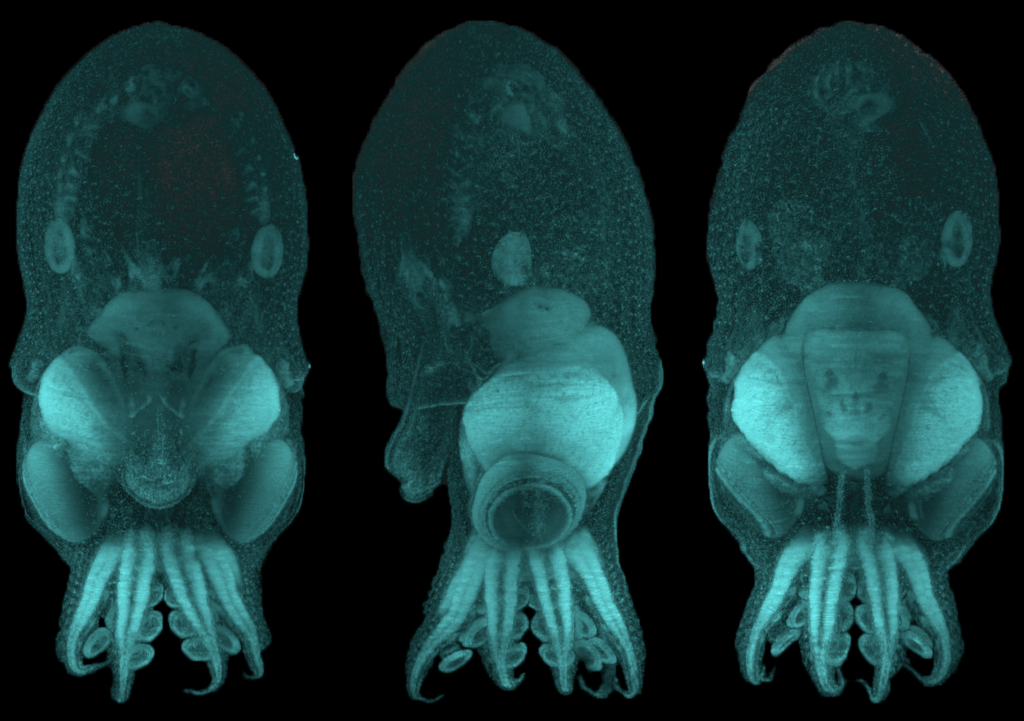
Despite the potential that the octopus provides for understanding developmental biology, particularly the brain, the molecules that dictate how the mollusk’s brain is built are unknown. The common octopus, Octopus vulgaris, is specifically suited to address this question because it can produce thousands of small and transparent eggs in a single batch and scientists have recently mapped out most of its genes. Using O. vulgaris in studies led by Dr. Astrid Deryckere from Dr. Eve Seuntjens’s lab at KU Leuven in Belgium, the group set out to unravel these molecular mysteries. “If you would think of cephalopods as the primates of the sea, that have evolved a complex nervous system from a far more simple ancestral nervous system, surprisingly little is understood on the morphological and molecular mechanisms driving its development.”, said Dr. Deryckere. They approached the problem in two studies. In the first study, she established a system for controlled embryonic development, which enabled her to care for thousands of eggs without the need for the mother octopus. She used state of the art microscopy and recorded high-resolution images of octopus development from fertilization through hatching. This work from Dr. Deryckere and colleagues can now be used as an elaborate reference for cephalopod embryology.
O. vulgaris hatchling imaged in 3D at high resolution after labelling DNA (cyan). The same embryo was imaged from different orientations: back view (left), side view (middle) and front view (right). The head is located on top and the arms are at the bottom. Images were produced by Dr. Astrid Deryckere in the lab of Dr. Eve Seuntjens, KU Leuven, Belgium.
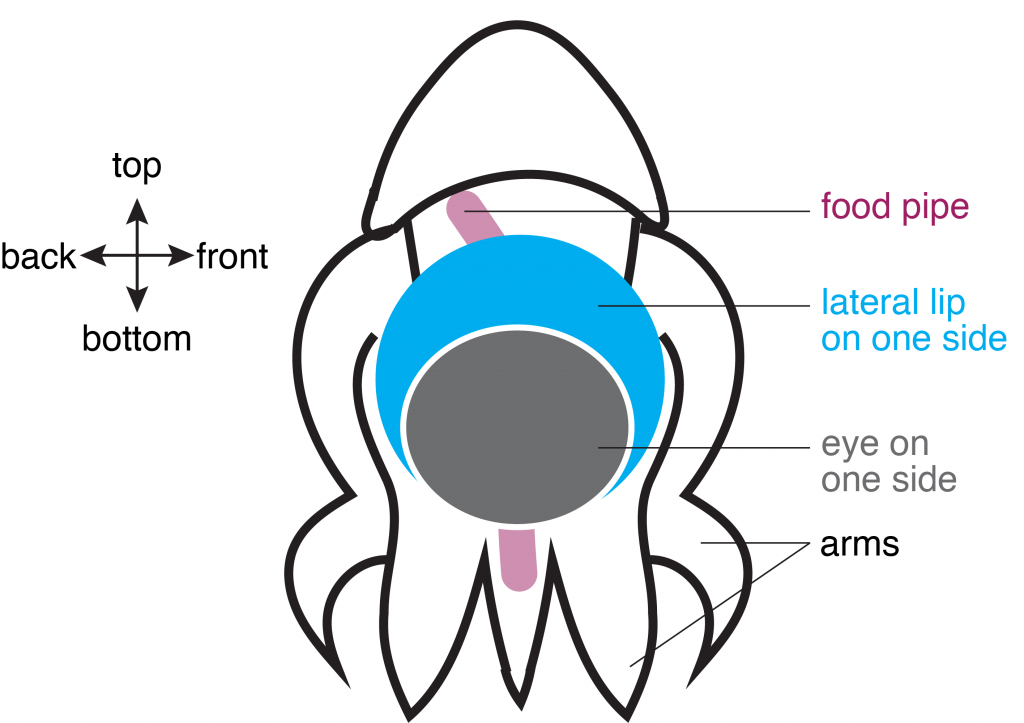
In the second study, Dr. Deryckere dug deep into the origins of the octopus brain. She used precise staging to track the precursor cells of neurons, or “neuronal progenitors”, that generate specialized neurons. Intriguingly, these progenitor cells appeared in structures called the lateral lips, that are unrelated to the brain and are located around the eyes. So, it appeared that neurons were first born in these structures and eventually migrated into the central brain – a possibility that prompted the authors to investigate further. They found that hundreds of thousands of neurons were created within octopus embryos even before hatching. To find out what genes are required for this unique way of making neurons, Dr. Deryckere used molecular markers and showed for the first time that newborn neurons travel long-distances to reach their final location in the brain. The results showed that the genes were expressed in the same order that vertebrates like humans do. By closely observing entire embryos in three dimensions during their growth, she found that neurons proceed through maturation while migrating from the lateral lips to an intermediate transition zone that finally leads to the brain.
Schematic of O. vulgaris embryo, indicating the location of the lateral lip in relation to the food pipe and the eye. Schematic adapted from images in Deryckere et al., 2021.
Using detailed molecular studies, the scientists now show support for the lateral lips harboring newly dividing neuronal cells in the embryo. This is unusual because unlike in human brains and many other organisms, the dividing cells are located outside the central brain. These dividing cells then unwaveringly make their way towards the final destination to maturity in the octopus central brain. “The migration is especially exceptional for invertebrates where neurons usually migrate only a few cell lengths”, noted Dr. Deryckere about the rarity of the observation.
This unique development in the octopus head and its interesting age-dependent arrangement of dividing cells and mature neurons only inspires further reverence for the cephalopod. While it continues to influence characters in pop culture, the glorious octopus and its brain hold even more promise in the real world. The octopus brain’s cognitive ability has galvanized a new age of artificial intelligence, leading to the construction of flexible robotics and prosthetics, but at the same time, is pushing scientists and philosophers to tackle the important question of how an intelligent life form is defined.
Dr. Astrid Deryckere is currently a postdoc in the lab of Maria Tosches in the Department of Biological Sciences at Columbia University. Her focus remains on brain development but she has transitioned to working on an animal with a backbone – the salamander.
Bacteria haven’t had much luck with the press. Since the advent of germ theory in the 1800s, pathogenic bacteria – the disease-causing “bad apples” of the bacterial barrel – have hogged the spotlight in both science and popular imagination. This trend has started shifting over the last decade, as researchers have turned their attention toward the human microbiome: the trillions of bacteria, archaea, and other microbes which reside inside the human body (primarily the gastrointestinal tract) and are now known to contribute to host immunity, metabolism, and even behavior.
But how? By what mechanisms do these microscopic guests communicate with their host to exert such profound effects? In contrast to well-characterized host-pathogen interactions, interactions between hosts and commensal (i.e., “friendly”) microbes have remained largely unexplored. In a recent collaborative study, Mark Ladinsky of the California Institute of Technology and Dr. Leandro Araujo of Columbia University sought to change that.
Ladinsky and Araujo focused their investigation on one class of microbes in particular: segmented filamentous bacteria (SFB), which make their home inside the small intestine of humans and other animals. Though these bacteria do not cause tissue damage or overt inflammation, they do stimulate an immune response in the host, resulting in the induction of immune cells that specifically recognize SFB and help control the population of these bacteria. This specificity relies on access of host cells to SFB antigens: foreign proteins or other distinctive components on bacterial targets that make them recognizable to immune cells. In the case of harmful bacteria, antigens arise in abundance as pathogens attack host cells and vice versa, but SFB don’t possess the machinery for invasion of host cells and show no hallmarks of destructive mechanisms. So how does the immune system come to recognize these peaceful residents? The answer, the scientists found, lies in a previously unknown communication pathway between commensal bacteria and host intestinal epithelial cells (IECs).
Using electron tomography, a technique that allows three-dimensional reconstruction of cellular and subcellular structures at high resolution, the authors found that IEC plasma membranes were forming small cavities exclusively where they interfaced with bacteria. These cavities eventually bud off into bubble-like vesicles inside the host cell. This series of events at the host-bacteria interface was characteristic of normal cellular processes for bringing external substances into cells, known as endocytosis. Upon discovering that the observed vesicles contained a bacterial cell wall protein and common SFB antigen, the researchers confirmed that this pathway – which they termed “microbial adhesion-triggered endocytosis,” or “MATE” – served as a means by which SFB make their presence known to their host. Thus, host IECs can sample their commensal bacterial population without consuming and destroying whole microbial cells. This peaceful transfer is likely advantageous in allowing the host to mount a mild immune response for SFB population control without triggering dramatic inflammation, though the mechanistic links between MATE and downstream immune effects remain unclear as of yet.
The authors, asking whether this unknown and surprisingly harmonious communication mechanism might be common among “healthy” microbes, next looked for signs of MATE among other classes of commensal intestinal bacteria, including those that are known to activate host immune responses similar to SFB responses. Though MATE communication was absent in all of the other species observed, the researchers noted that none of these microbes associated directly with IECs, as they observed in the case of SFB. Indeed, apart from SFB, the only microbes known to interact closely with IECs are bacterial pathogens, which themselves showed no signs of MATE signaling. These findings might indicate that MATE is a unique communication method specifically between host IECs and SFB (or other, as-yet-unidentified bacterial species), but they also suggest that strategies for crosstalk between microbes and hosts may be as diverse as the microbes themselves.
Like MATE, many new pathways of host-commensal interaction might be awaiting discovery. Such pathways could someday open doors for alternative vaccine or drug-delivery strategies, reducing the necessity for much-dreaded needle shots. They may even facilitate therapies for regulating microbial populations as a revolutionary treatment for metabolic diseases like irritable bowel syndrome or obesity. If so, perhaps “germs” might get a little credit as heroes in the story of human health. Some good press at last.
Mark Ladinsky is an Electron Microscopy Scientist at the California Institute of Technology. Dr. Leandro Araujo is a Postdoctoral Research Scientist in the Department of Microbiology & Immunology at Columbia University.
Around 18 million people die from cardiovascular disease each year, making it the leading cause of death worldwide. The main cause of cardiovascular disease is atherosclerosis, a process that occurs when fatty substances, cholesterol, and cell debris accumulate in blood vessel walls and form so-called “atherosclerotic plaques”. The progressive development of atherosclerosis is complex, as it involves genetic predispositions as well as environmental factors, such as an unhealthy diet, physical inactivity and smoking. Over time, atherosclerotic plaques can become unstable and prone to rupture. Plaque rupture leads to the formation of a blot clot or “thrombus”, which can occlude a blood vessel and thereby cause a heart attack or stroke.
Various cell types in the blood vessel wall contribute to the initiation and progression of atherosclerosis, including smooth muscle cells (a type of muscle cell found in the walls of hollow organs), endothelial cells (that line the inner surface of blood vessels), and macrophages (a large immune cell found in tissues at sites of infection or tissue damage). Remarkably, smooth muscle cells can change the way they look and function depending on the tissue microenvironment, a process referred to as “phenotypic switching”. Dr. Huize Pan, a postdoc from Columbia University, investigated this phenomenon in the context of atherosclerosis, to find out whether smooth muscle cells are the good or bad guy in this disease. As it turns out, they are both.
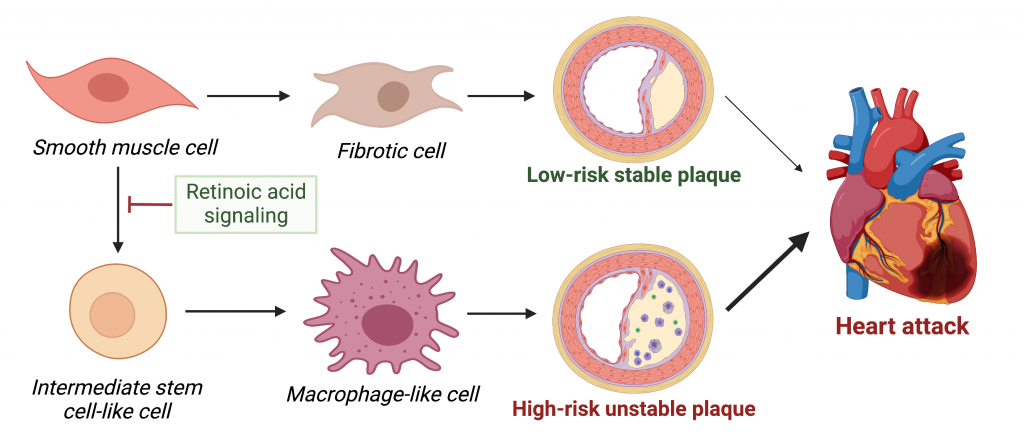
Smooth muscle cells can transition to fibrotic cells that synthesize a fibrous cap covering the atherosclerotic plaque. This is a beneficial process as it reduces the likelihood of plaque rupture. However, in their recent paper published in Circulation, Dr. Pan and colleagues show that smooth muscle cells can also turn into intermediate stem cell-like cells that can further differentiate into macrophage-like cells (see Figure below). This “shapeshifting” of smooth muscle cells towards macrophage-like cells could be harmful as certain macrophages are known to promote plaque inflammation and instability.
Depending on the state of retinoic acid signaling, smooth muscle cells can either turn into fibrotic cells and play a protective role in atherosclerosis, or turn into intermediate stem-cell like cells that give rise to inflammatory macrophage-like cells, thereby increasing plaque instability and the risk of heart disease. Figure adapted from Pan, Circ 2020, and created with BioRender.com.
This raises the question of how smooth muscle cells either become more fibrotic and play a protective role in atherosclerosis or transdifferentiate into inflammatory macrophage-like cells and play a damaging role. Through single-cell RNA sequencing analysis, a research method to examine which genes are turned “on” and “off” in individual cells, Dr. Pan and colleagues found significant differences in target genes of retinoic acid signaling between smooth muscle cells and intermediate stem cell-like cells. This indicates that signaling through retinoic acid, a derivative of vitamin A that helps regulate growth and development, could be an important mechanism by which smooth muscle cells transition to other cell states (as depicted in the Figure above).
Next, the researchers explored whether these findings are relevant for human heart disease. Indeed, they found dysregulated retinoic acid signaling in human atherosclerotic plaques, and discovered that human individuals with genetic variation in target genes of retinoic acid signaling have a higher risk of cardiovascular disease. These findings suggest that by determining smooth muscle cell fate, retinoic acid signaling controls the outcome of atherosclerotic cardiovascular disease. Manipulation of retinoic acid signaling could therefore be a promising therapeutic strategy to reduce cardiovascular risk. This is supported by the current study, through the use of an FDA-approved drug named ATRA (activation of RA signaling by all-trans RA) that activates retinoic acid signaling. ATRA reduced the number of smooth muscle cell-derived macrophages, reduced atherosclerosis progression, and increased fibrous cap thickness in a mouse model of atherosclerosis.
Taken together, these novel findings indicate that smooth muscle cells can play both the good and bad guy in atherosclerosis. By promoting smooth muscle cells in atherosclerotic plaques to follow the “righteous path”, we are one step closer to a world free of heart disease.
Dancing can be one of life’s greatest pleasures. But for folks who consistently engage in intensive forms of dance, such as ballet, it can also lead to injury. One injury amongst dancers and other athletes is flexor hallucis longus tendinopathy.
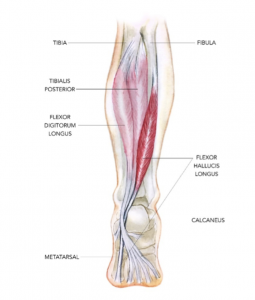
The flexor hallucis longus tendon (FHL), as seen in Figure 1, helps stabilize a person when they’re on their toes and mainly moves to flex the big toe. It stretches all the way from the calf muscle, through the ankle, down to the big toe. When athletes engage in repetitive movements that recruit their foot and ankle in this manner, like jumping up and pushing off the big toe, strain of the FHL tendon can occur. FHL tendinopathy is painful and can leave dancers and gymnasts out of commission from their passion and profession.
Figure 1. The posterior view of the FHL in the right leg, taken from Sports Injury Bulletin.
Luckily, researchers study overuse conditions like this one. Dr. Hai-Jung Steffi Shih and her colleagues recently published a study where they had 17 female dancers (9 with FHL tendinopathy and 8 without) perform a specific ballet move called saut du chat. The dancers wore a full-body marker set that allowed the researchers to capture the fine-grained positions and movements of the dancers’ bodies throughout the ballet move.
When performing a movement like the saut du chat, the body tends to place a heavy load on one particular joint in the foot called the metatarsophalangeal joint (MTP). Repeating the movement like this over and over again (as dancers often do), can contribute to overuse of the FHL. Researchers can measure something called stiffness in an athlete’s musculoskeletal system to assess the potential for injury. Scientists think that greater stiffness may lead to impact on a person’s bone, while reduced stiffness may lead to soft tissue injury. To better understand stiffness, it can help to think about parts of the lower body as springs, such as the ankle, knee, and hip joints. They compress, store energy, and then release – like when you squat and jump. Researchers can examine stiffness of these joints, specifically joint torsional stiffness, or how easy or hard the joints bend.
Additionally, researchers who study movement can also measure how a dancer’s body makes contact with the ground to determine if certain kinetic factors might be significantly associated with injury. For instance, the difference in the angle at which a dancer’s lower limb makes contact with the ground has been associated with injured and uninjured groups of dancers. If researchers can accurately indicate which angles are associated with injury, they can collaborate with medical professionals and teachers to craft targeted interventions on how a dancer should be properly moving their body.
In the current study, the researchers posited that lower extremity joint torsional stiffness measured when participants made contact with the ground would be altered in dancers with tendinopathy compared to uninjured dancers. They also thought that dancers with tendinopathy would demonstrate a lower limb posture associated with kinetic factors that differed between injured and uninjured dancers.
Using the marker set that the dancers wore, the researchers measured torsional stiffness by examining the rotational forces, or the change of joint movements, over the change of the joint angle over a period of time. This data was gathered as the dancers flexed and extended their lower extremity joints during the saut du chat. Moreover, the team measured the contact angle at which the dancers’ feet took off from.
Dr. Shih and her colleagues found that the dancers with tendinopathy demonstrated less joint torsional stiffness at the metatarsophalangeal (MTP), ankle, and knee joints during the takeoff step of the dance move. To reiterate, research suggests that a lack of joint stiffness is not good because it could lead to soft tissue injury, as it allows for excessive joint motion. Additionally, these injured dancers took longer to reach peak force when pushing off the ground and the peak force was also lower than in the uninjured dancers. Finally, the angle at which dancers first contacted the ground during that take off step was smaller (i.e., their foot was further in front of their pelvis) in participants with FHL tendinopathy compared to those without injury.
How can these findings help dancers and those that provide movement guidance to dancers? Knowing the particular biomechanical changes that precede tendinopathy can inform targeted interventions aimed at improving how dancers move their feet and legs when performing certain moves. For example, teachers can offer cues and guidance on how a dancer should position their pelvis and feet in an effort to prevent injury.
This study by Dr. Shih and her colleagues is the first to demonstrate the differences in movement profiles between dancers with and without FHL tendinopathy and could go a long way to informing interventions that could prolong dancers’ careers.
Dr. Hai-Jung Steffi Shih is currently a postdoctoral research fellow in the Neurorehabilitation Research Lab at Teachers College, Columbia University. She received her PhD in Biokinesiology and Physical Therapy at the University of Southern California where this research was conducted. Steffi’s research aims are to further understand musculoskeletal pain, movement disorders, and improve intervention strategies using a multidisciplinary approach. Outside of academia, Steffi is an avid traveler who has been to more than 35 countries. She loves to dance, enjoys playing music, and is aspiring to become an excellent dog owner one day. You can email her at [email protected] and connect with her on Twitter @HiSteffiPT.
When Dr. Jaan Altosaar heard that food deprivation increases stem cell regeneration and immune system activity in rats, he did what many would not dare: he decided to try it himself and fasted for five days. Thoughts of food started to take over his mind and, with what can only be qualified as a superhuman ability to think with low blood sugar, he went on a scientific tangent and channeled them into tackling the complicated task of improving foodrecommendation systems,which led to publishing a research article about it.
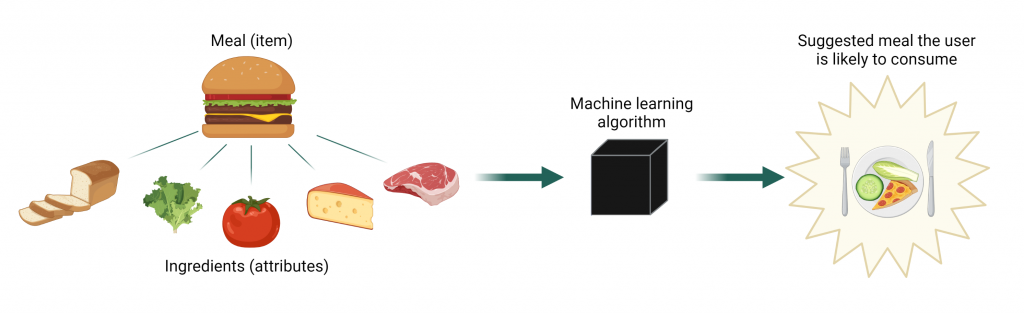
Dr. Altosaar wanted help in making decisions because choosing is hard. When faced with an excessive number of options, we fall victim to decision fatigue and tend to prefer familiar things. Companies know this, and many have developed personalized recommendations for many facets of our lives: Facebook’s posts on your timeline, potential partners on dating apps, or suggested products on Amazon. But Jaan had a clear favorite: Spotify’s Discover Weekly algorithm. The music app gathers information on co-occurrence of artists in playlists and compares the representation of you as a listener to the couple billion playlists it has at its disposal to suggest songs you might enjoy. Since Dr. Altosaar’s problem was similar, he framed the problem as feeding the algorithm a user’s favorite recipes (“playlists”), which are made of a list of ingredients (“songs”). Would the algorithm then cook up suggestions of complimentary meals based on the ingredients in them?
A meal consumed by a user (hamburger) is made up of ingredients (bread, lettuce, tomato, cheese, meat). This information is given to the machine learning algorithm, which will use learnt information about that user to provide a recommendation likely to be eaten by them.
Meal recommendation in an app is challenging on several fronts. First, a food tracking app might record eating the same meal in many different ways or with unique variations (such as a sandwich with homemade hot sauce or omitting pickles). This means that any specific meal is typically only logged by a small number of users. Further, the database of all possible meals a user might track is enormous, and each meal only contains a few ingredients.
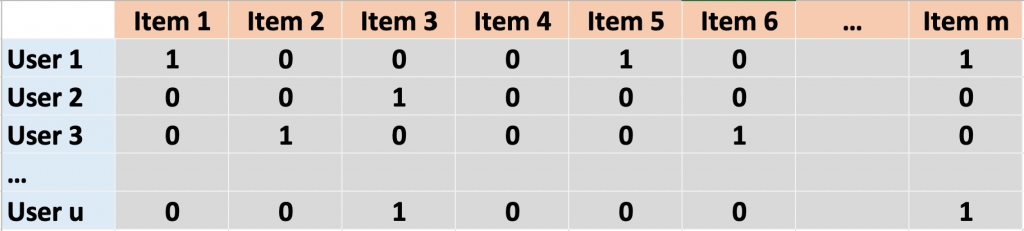
In traditional recommender systems such as those used by Netflix, solving this problem might mean first translating the data into a large matrix where users are rows and items (e.g. movies or meals) are columns. The values in the matrix are ones or zeros depending on whether the user consumed the item or not. Modern versions of recommender systems, including the one in Dr. Altosaar’s paper, also incorporate item attributes (ingredients, availability, popularity) and use them as additional information to better tailor recommendations. An outstanding issue, however, is striking a balance between flexibility, to account for the fact that we are not all like Joey Tribbiani and might not like custard, jam and beef all together (even if we like them separately), and scalability, since an increasing number of attributes takes a toll on computing time. Additionally, these machine learning algorithms are not always trained the same way they are later evaluated for performance.
A sparse matrix representing whether a user “u” consumed an item “m” (coded with a one). If the user did not consume the item, there is a zero. Note that most items in the matrix are zeroes, so that there is not a lot of actual information (thus calling it sparse).
The new type of model Dr. Altosaar and colleagues propose, RankFromSets, frames the problem as a binary classification. This means that it learns to assign a zero to meals unlikely to be consumed by a user, and a one to those that are likely to be consumed. When faced with giving a user a set of potential meals (say five), it strives to maximize the number of meals the user will actually eat from those five recommended to them. To leverage the power of incorporating the meal’s ingredients, the algorithm uses a technique from natural language processing to learn embeddings. These are a way to compress data to preserve the relevant information you care about to solve your problem; in this case, learning about patterns useful for predicting which ingredients tip the balance for someone to consume a meal. This allows for a numerical representation for each meal based on its constituent foods, and the patterns in how those foods are consumed across all users.
The RankFromSets classification model incorporates several components. There are embeddings for representing user preferences alongside the embeddings corresponding to a meal a user might consume. The classifier is spiced up with additional user-independent information about the meal’s popularity and its availability. These components are used by the model to learn the probability that a particular meal will be consumed by a user. Potential meals a user might enjoy – or that might be healthier options – are then ranked, and the top meals will be recommended to the user. For example, if you have had avocados in every one of your meals, they are in season, and all those Millennials are logging in their avocado toast, you are very likely to receive recommendations that include avocados in the future.
As a proof of concept, the authors tested their method not only on food data, which they got from the LoseIt! weight loss app, but also on a dataset unrelated to meal choices. For this independent data set, the authors used reading choices and behavior among users of arXiv, a preprint server. They trained the model on past user behavior data and evaluated performance (accuracy of paper suggestions) on a previously separated portion of that same data (so they knew whether the user had actually read the paper, but this information was hidden from the algorithm for evaluation). This is a typical way to assess the performance of machine learning systems, and their method outperformed previously-developed recommender systems. The better performance and translatability to tasks other than meal recommendation is indicative of the potential of this tool to be applied in other contexts.
This new recommender system could be applied to either recipe recommendation apps, or even to an app that would suggest first-time customers of a restaurant the menu items that they are more likely to like based on their preferences. The system also has the potential to incorporate additional information beyond whether a user consumed (and liked) a particular ingredient or meal. Sometimes the method of cooking determines whether a food is appealing or not (Brussel sprouts I’m looking at you). Additionally, classifying ingredients by flavor might also be helpful in suggesting similar (and even healthier) alternatives. Therefore, adding those tags as extra layers to the user-meal intersection will certainly provide better recommendations and opportunities to cook outside of the box. Dr. Altosaar’s fast might or might not have gotten him a boost in his stem cells, but he certainly succeeded in helping everyone else worry a bit less about what’s for dinner tonight.
Dr. Jaan Altosaar is a Postdoctoral Research Scientist in the Department of Biomedical Informatics and an active participant in CUPS. He publishes an awesome blog about machine learning and its implications.
The human body has approximately 15 trillion cells, all of which arise from embryonic stem cells which are considered the building blocks of life. Stem cells renew themselves by dividing indefinitely and can also give rise to cells with specialized functions which ultimately end up forming various organs and tissues in our body. This process is called differentiation. Typically, once cells specialize or differentiate, they lose the ability within the body to go back to being stem cells. Given their unique properties, stem cells have become a critical starting point that scientists can tinker with to develop new drugs and therapies. Because of their tremendous value for research, scientists have figured out non-invasive ways to transform differentiated cells into cells with stem cell like properties. These lab-grown cells, called induced pluripotent stem cells or iPSCs, are typically generated by a process called “cellular reprogramming”.
As Dr. Tania Thimraj explains in a recent article, proteins called transcription factors can act as cellular “fixer-uppers” and renovate differentiated cells to look and behave like stem cells. The current state of the art process for making iPSCs involves excess production, also known as overexpression, of the following transcription factors in differentiated cells: Oct4, Sox2, Klf4, and c-Myc (collectively called the “OKSM” cocktail). Despite significant advances in the formulation of this cocktail, there is still a huge margin for improvement in the ability of this cocktail to transform differentiated cells into stem cells. In a recent study performed by Dr. Tan and co-authored by Dr. Malik, the authors propose that the cocktail is not especially effective because the transcription factors were never under any evolutionary selection pressure to produce stem cells. Inspired by this, the authors set out to use evolution in the dish, also known as directed evolution, to make a more efficacious transcription factor cocktail.
Although natural evolution takes place over millions of years, smaller scale evolution can be done in a laboratory setting at much faster timescales. This approach is known as “directed evolution” and has been successfully used by scientists to evolve proteins with new functionalities. This process involves making random mutations in the protein of interest. Then, these mutants undergo a selection process in an appropriate cellular context so that protein variants with desirable properties can be isolated.
In a pioneering study, members of the Jauch lab, including Dr. Malik, used directed evolution to optimize the cellular reprogramming ability of the transcription factor Sox2. Building on this success, the Jauch lab used directed evolution to make ePOU, an enhanced and evolved version of Oct4 which is an integral part of the OKSM cocktail. In the current study for creating ePOU, the authors made random mutations at six functionally important positions in Oct4 and overexpressed the mutant proteins in mammalian cells such that the Oct4 transcription factor activity was tied to the production of a green fluorescent protein representing stem cell transformation.
This innovative study demonstrates that the transformation potential of naturally occurring transcription factors can be drastically enhanced by directed evolution. In addition, this work also provides a framework for future research on transcription factor engineering for cell reprogramming. By providing a faster and more efficient way to produce stem cells, this study has the potential to accelerate various research and therapeutic avenues such as regenerative medicine, drug efficacy and safety testing, and studying human development and disease.
Dr. Vikas Malik is a Postdoctoral Research Fellow in Dr. Jianlong Wang’s lab in the Department of Medicine at Columbia University Medical Center and is a member of CUPS and the Outreach and Communications Committee.
For some people, this statement is absolutely invalid as they never feel full; they don’t have an ‘off-switch’ while eating. Sometimes, consuming food makes them feel even hungrier. These are some classic symptoms of binge eating. Binge eating falls under the big umbrella of eating disorders, which are serious mental health conditions characterized by persistent alteration of eating behavior and associated emotions. Three different diseases belong to the spectrum of eating disorders: anorexia nervosa, bulimia nervosa, and binge eating disorders. Although binge eating disorder is the most prevalent, surprisingly it does not get as much media coverage compared to anorexia and bulimia.
Binge eating results from “hedonic hunger” the drive to consume food not because of an energy deficit, but for the inherent pleasure associated with eating. The pleasure signal for bingeing relies mostly on the reward-associated component of feeding and sensory stimuli such as smell and taste. The reward system functions by raising the level of the neurotransmitter dopamine in a midbrain structure called the ventral tegmental area. Years of research in laboratory animals also depicted a positive correlation between binge eating and increased dopamine release. The endocannabinoid system has been connected with this rewarding aspect of food intake and represents the key system modulating bingeing. Fun fact, cannabis consumption leads to overeating (read: munching) by tricking the brain into feeling like it’s starving when in reality that’s not the case. In association with the endocannabinoids and reward system, the gut or gastric lumen also plays as a master driver controlling feeding behavior in general along with binge eating. Intriguingly, endocannabinoids are functionally dependent on the vagus nerve innervating the gastrointestinal tract. Overall, scientists have just begun to understand the complex nature of binge eating from the neurobiological and psychological standpoint.
A recent preprint by Dr. Chloé Berland and colleagues dissected, for the first time, the indelible role of the reward system, gut-brain axis, and endocannabinoids in binge eating. This study successfully leveraged a unique binge eating model in which a highly palatable milkshake was provided to mice in a time-locked manner. This binge-eating model was driven by reward values rather than metabolic demand, as animals had unlimited access to less palatable food throughout the test, so milkshake consumption occurred in absence of energy depreciation. This study pinpointed that two phases of binge eating, anticipatory and consummatory, are controlled by a specific dopamine receptor called D1 (D1R).
The cannabinoid receptors are available both in the peripheral and central nervous systems. The current study aimed to uncover the specific connection between peripheral cannabinoid and bingeing. To achieve that goal, a peripherally restricted chemical was administered in the mice to block the activity of the cannabinoid receptor. Dr. Berland and her colleagues observed that the injection of peripheral cannabinoid blocker completely silenced the hedonic drive for bingeing. This finding reveals that physiologically, the peripheral endogenous cannabinoid acts as a gatekeeper for binge eating.
Figure 1. Schematic representation showing how the peripheral endocannabinoid mediates bingeing via the gut-brain axis. The left panel of the diagram shows that increased peripheral endocannabinoid causes increased reward and bingeing while the right side shows the opposite. The brain region, Nucleus Tractus Solitarius (NTS), Endocannabinoids, or eCB (2-Arachidonoylglycerol molecule as a representation). Adapted fromBerland et al. and created with Biorender.com.
To delve more into the involvement of the gut-brain axis in endocannabinoid-mediated bingeing, the current study used vagotomy, a severing of the vagus nerve’s connections to the gastrointestinal tract and other abdominal organs, to shut off the function of the vagus nerve in these organs. Injection of peripheral cannabinoid blocker in vagotomized mice led to strong activation of a brain region known to play a key role in receiving signals from the gut about meals, the nucleus tractus solitarius (NTS) (see Figure 1). This observation indicates that the peripheral endocannabinoids are important influencers that act in between the gut and brain in regulating the hedonic drive for food.
This study took advantage of a unique, cutting-edge technology called fiber photometry to further dissect how the endocannabinoids control the reward component of bingeing. With fiber photometry, the neural activity of specific brain regions can be detected in awake animals. The neural activity in the midbrain reward area was dampened after the peripheral endocannabinoid blocker injection. This finding suggests that peripheral endocannabinoids control the food craving by modulating the reward system.
Taken together, the observations of this study provide crucial mechanistic insights on gut-brain and endocannabinoid integration. Using state-of-the-art tools, this study sheds light on the previously unexplored regulatory mechanism of the endocannabinoids in bingeing. So, the next time you binge eat a pint of Ben & Jerry’s ice cream, you know it’s not only the burst of pleasure chemical dopamine but also your body endocannabinoids tricking your gut and brain to finish it all.
These new and exciting data warrant that peripheral endocannabinoid blockers could be utilized for the treatment of binge eating disorders or related eating disorders in humans. Patients with eating disorders struggle mentally, emotionally, and physically. For instance, individuals with eating disorders often become victims of body shaming. We can always do more to help binge eating disorder patients in the recovery process. Here are some useful resources for patients struggling with binge eating disorder:
Dr. Chloé Berland is a Postdoctoral Research Scientist in the Department of Preventive Medicine where she studies the effect of overfeeding on brain circuits. She also serves as CUPS secretary.