Have you ever caved in to the temptation of a snack in the middle of the night that manifested into a quick freezer dive to grab that ice-cream or into a series of quick taps on your food delivery app to get those udon noodles? Suffice it to say that I have been a victim to this thought one too many times. Much to my chagrin, there is an abundance of evidence that suggests that eating during restricted hours of the day or time-restricted feeding (TRF) can slow down decline of bodily functions. Limiting food intake to certain hours of daytime, even if the food is not necessarily nutritious or low in calories, can prevent ageing or even kickstart anti-aging mechanisms in mice and flies with obesity or heart disease. Because ageing was dependent on when the body takes in food, these studies hint at the role of the body’s biological clock, known as circadian rhythms, in regulating health and longevity. In an unexpected new study authored by Columbia postdoc Dr. Matt Ulgherait, flies following time-restricted feeding while also balancing it with an unlimited all-access ad libitum diet, show a significant increase in lifespan.
By structuring 24 hour day-night periods as cycles of 12 hours of light followed by 12 hours of darkness in a temperature-controlled box, the authors tested various dietary regimens for their effects on lifespan and stumbled upon one regimen that consistently showed longer lifespan along with enhanced health in the flies. This regimen cycled between a 20-hour fast starting at mid-morning (6 hours after lights on) to a 22 hour recovery period of eating ad-libitum on repeat in young flies within 10-40 day post hatching stage of adulthood. However, flies that began this regimen after reaching older age at day 40 did not show enhanced lifespan. In comparison to flies that were allowed access to food ad-libitum on a 24 hour cycle, flies following this particular fasting-feeding regimen showed a 18% increase in female lifespan and 13% increase male lifespan in their young age. Due to the cycling schedule of unlimited food access with periods of fasting, the authors termed this regimen as intermediate time-restricted feeding (iTRF).
Previous studies have shown that caloric restriction through reduced food intake, protein restriction or inhibiting insulin-like signaling can extend lifespan. However, iTRF did not appear to limit flies from eating less and in many cases, resulted in flies eating more during times of food access compared to those in the ad libitum group. Thus, lifespan extension under iTRF did not occur because of limitation in nutrient uptake. Interestingly, an iTRF regimen performed under additional treatments of either dietary protein restriction or inhibited insulin-like signaling, resulted in a marked boost in lifespan compared to iTRF alone. It therefore seems that independent mechanisms that can enhance lifespan can be combined to increase lifespan even more.
While these methods provide ways to extend lifespan through incremental means, some might argue that it would be meaningless to simply survive without long-lasting health benefits. To examine whether the longer-lived flies continued to exhibit youth, scientists measured the fitness of the flies using two well-known age-related tests: the flies’ ability to climb up the plastic vial they are in and how much they accumulate in their tissues aggregates of aging proteins – polyubiquitin and p62. When compared to the ad libitum group, iTRF flies climbed much faster and had fewer polyubiquitin and p62 aggregates in the flight muscles, even after they reached an age beyond 40 days of hatching. While the gut microbiome was shown to dictate proclivity for disease and thus have an effect on lifespan, the gut tissue in iTRF flies remained healthier with more normal cells, even when the gut microbiome was depleted with antibiotics. Therefore, the flies appeared to be in optimal health conditions with fewer aging markers in addition to longer survival, demonstrating yet again that aging slowed down due to better functioning of organs.
The dietary regimen under iTRF only controls the timing of feeding but not the nutritional intake, which provided clues to the authors that perhaps the body’s natural biological clock had something to do with iTRF-mediated lifespan. The biological clock in flies consists of proteins that are also present in other organisms all the way from fungi to humans. The main molecular parts of the core circadian clock include the proteins ‘Clock’ (Clk) and ‘Cycle’ (Cyc) which activate the genes period (per) and timeless (tim), which in turn inhibit Clk and Cyc. This process is called a feedback loop which takes all of 24 hours to complete in both flies and humans, and this is how our bodies respond to light-dark cycles. Flies undergoing iTRF showed enhanced expression of Clk in the daytime and of per and tim at night time. The authors then explored the feeding behavior and metabolism of circadian clock genetic mutants undergoing iTRF and found that neither the 20 hour long fasting period nor dietary restriction in their food altered their feeding behavior when compared to normal flies under iTRF. Yet, the extended lifespan was completely missing in Clk, per and tim mutants undergoing iTRF. Even the improved health seen with an iTRF regimen through better climbing ability and less aging-protein aggregation was abolished in per mutants compared to normal flies. Shifting the iTRF cycle by 12 hours with a fasting period during the daytime abolished the occurrence of an extended lifespan. In the altered regimen, while the same cycle was now only shifted by half a day, eating at night time while fasting during the day just did not work. This discovery showed that there could be a deep link between the body’s biological timer and when during the day food is eaten that determines both longevity and well-being.
Because shifting the fasting period to daytime did not show any benefits, the authors checked whether genes that activate during fasting are also linked to the biological clock. In fact, Dr. Ulgherait and group had already shown that disrupting tim and per genes in the gut, which is where food is processed, caused an increase in lifespan. But, iTRF included periods of starvation that could trigger different metabolic processes. Starvation induces cellular mechanisms to degrade and recycle its molecules in a process called autophagy. Interestingly, genes encoding two autophagy proteins, Atg1 and Atg8a, which are also present in humans, showed peak levels in the night time with enhanced peaks in flies under iTRF. During autophagy, there is an increased activity of cell organelles called lysosomes that contain digestive enzymes needed to break down cellular parts. The authors found that normal flies fasting under iTRF showed higher Atg1 and Atg8a expression along with more lysosomal activity but period mutants failed to do so. Using some more genetic tricks, the authors found that manipulating the level of autophagy to go up or down directly showed an effect on iTRF-mediated lifespan.
Finally, to explore the link between iTRF-mediated lifespan and autophagy, the authors used genetic tools to increase night-specific levels of Atg1 and Atg8a. In a surprising revelation, flies with night-specific expression of Atg1 and Atg8a showed an increase in lifespan, even when these flies did not undergo fasting and were fed ad libitum. Subjecting these genetically altered flies to iTRF did not additionally increase their lifespan, suggesting to the authors that circadian enhancement of cellular degradation under an all-access diet provides the same beneficial effects as fasting done under the stricter regimen of iTRF. Flies with night-specific enhanced autophagy also showed better neuromuscular and gut health on an all-access diet. Therefore, clock-dependent enhancement of the biological recycling machinery can mimic the lifespan extension mediated by iTRF.
Now of course large genetic manipulations are not yet a consideration in humans but this study provides a potentially powerful yet simple change in dietary strategy that could just somehow slow down aging. Aging increases risk of mortality and disease but imagine a food intake regimen translatable from this study into humans that can help improve overall neuromuscular and gut health. So, while technology has indeed made it so much easier than before to have food at our doorstep in a few phone taps in the middle of the night, perhaps restricting the hours of when we eat can really help us live healthier lives. This study now makes me reconsider the famous quote by Woody Allen in the context of food, “You can live to be a hundred if you give up all the things that make you want to live to be a hundred”.
Dr. Matt Ulgherait is a postdoctoral researcher in the lab of Dr. Mimi Shirasu-Hiza in the department of Genetics & Development at Columbia University. Dr. Ulgherait and his colleagues also recently showed that removing the expression of the period gene from the gut tissue was sufficient to cause an increase in lifespan.


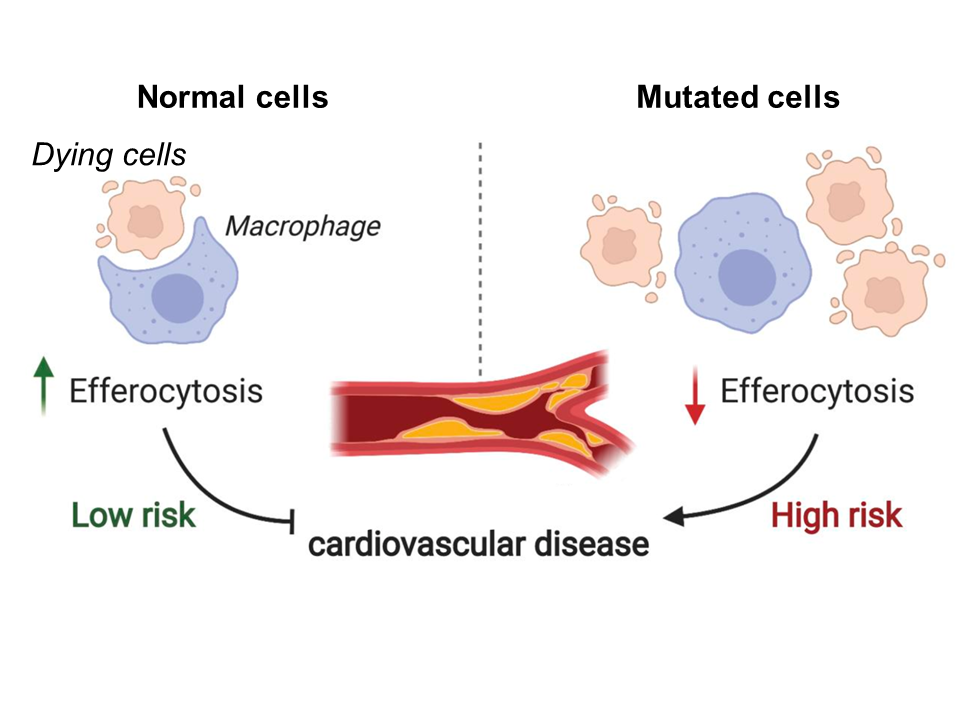 Fig 1. Model depicting the relationship between efferocytosis and risk of coronary artery disease. Reduced levels of efferocytosis lead to insufficient clearance of dead cells and consequent plaque formation in the arteries. Figure adapted from
Fig 1. Model depicting the relationship between efferocytosis and risk of coronary artery disease. Reduced levels of efferocytosis lead to insufficient clearance of dead cells and consequent plaque formation in the arteries. Figure adapted from 



.svg&oldid=468464751){kind=link}
{kind=link}