Many new simulation techniques such as Molecular Dynamics (MD), Dissipative Particle Dynamics (DPD), Coarse Grained Molecular Dynamics (CGMD) and Stochastic Rotation Dynamics (SRD) have been developed during the past few years to understand and predict molecular and mesoscale phenomena for systems such as polymers, colloids, and soft materials. A unique advantage of simulation methods is that molecular level details can be obtained about a given system for a particular application. We have initiated a program to utilize some of these simulation techniques to enhance our understanding of relevant problems such as adsorption, dispersion, and wetting etc.
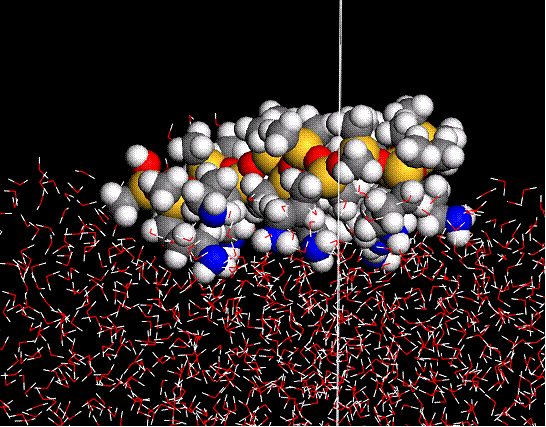

Aim of this program is twofold. The first objective is to understand the dynamics at atomic levels, which are difficult to characterize using experiments with an acceptable degree of confidence. The second aim is to explain the experimental findings through a rational modeling framework that is verifiable through subsequent simulations. For example, we have used Molecular Dynamics simulations using compass forcefield method to calculate the preferred conformations and orientations of hybrid silicone polymers at air-water interface as a function of nature of functional modifications and surface concentrations. At low concentrations, poly(dimethylsiloxane) was observed to be in a stretched conformation, whereas acid and amino modified siloxane preferred a serpentine shape. With an increase in concentration, all of the polymers oriented themselves in coiled conformations, with the ionic ones immersing deeper into the web such that the interactions among functional groups and water are maximized. Understanding the fundamental properties the proposed approach can help in tailoring the reagents for specific applications.
